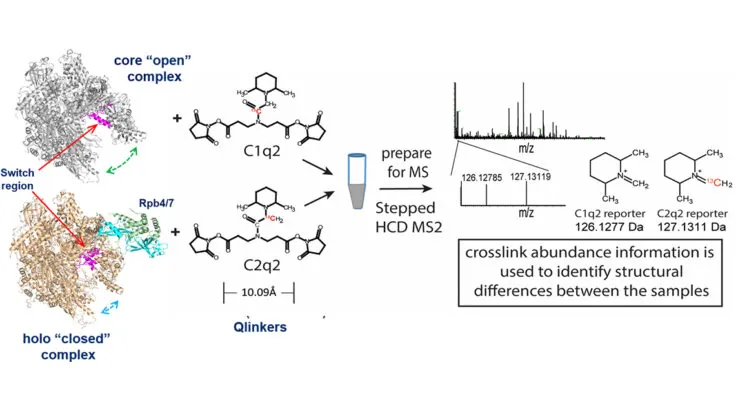
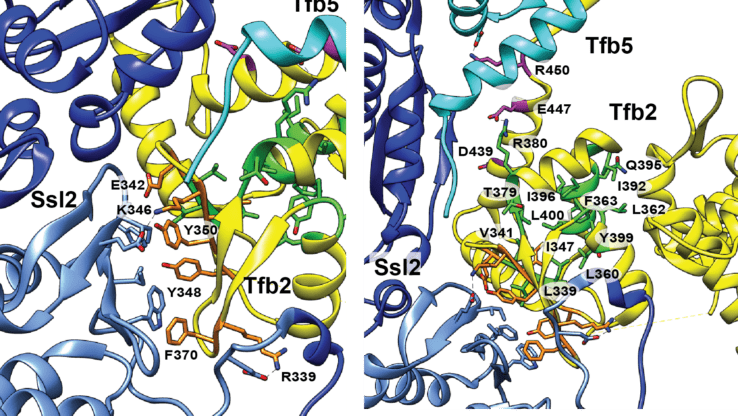
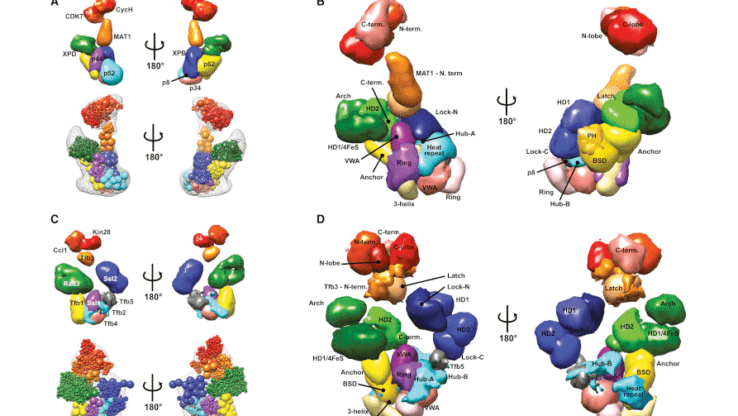
A new crosslinking mass spectrometry technology for studying conformational and structural changes in protein complexes
Our work describing a new crosslinking mass spectrometry technology for studying conformational and structural changes in proteins and proteins complexes has been published in eLife.